Familiaalinen hyperkolesterolemia (FH)
FH on periytyvä eli geneettinen sairaus.
Familiaalisella hyperkolesterolemialla (FH:lla) on erityisasema kolesterolihäiriöiden joukossa, koska se on elinikäinen, vaikea-asteinen, aiheuttaa suurella todennäköisyydellä varhaisen valtimotaudin ja on ajoissa todettaessa hoidettavissa.
- Mikä nostaa kolesterolia FH:ssa?
- FH on periytyvä eli geneettinen sairaus
- FH on kolesterolitauti
- FH altistaa valtimosairaudelle
- Miten FH todetaan?
- Geenitesti
- Suvun tutkiminen
- FH:n hoito
Mikä nostaa kolesterolia FH:ssa?
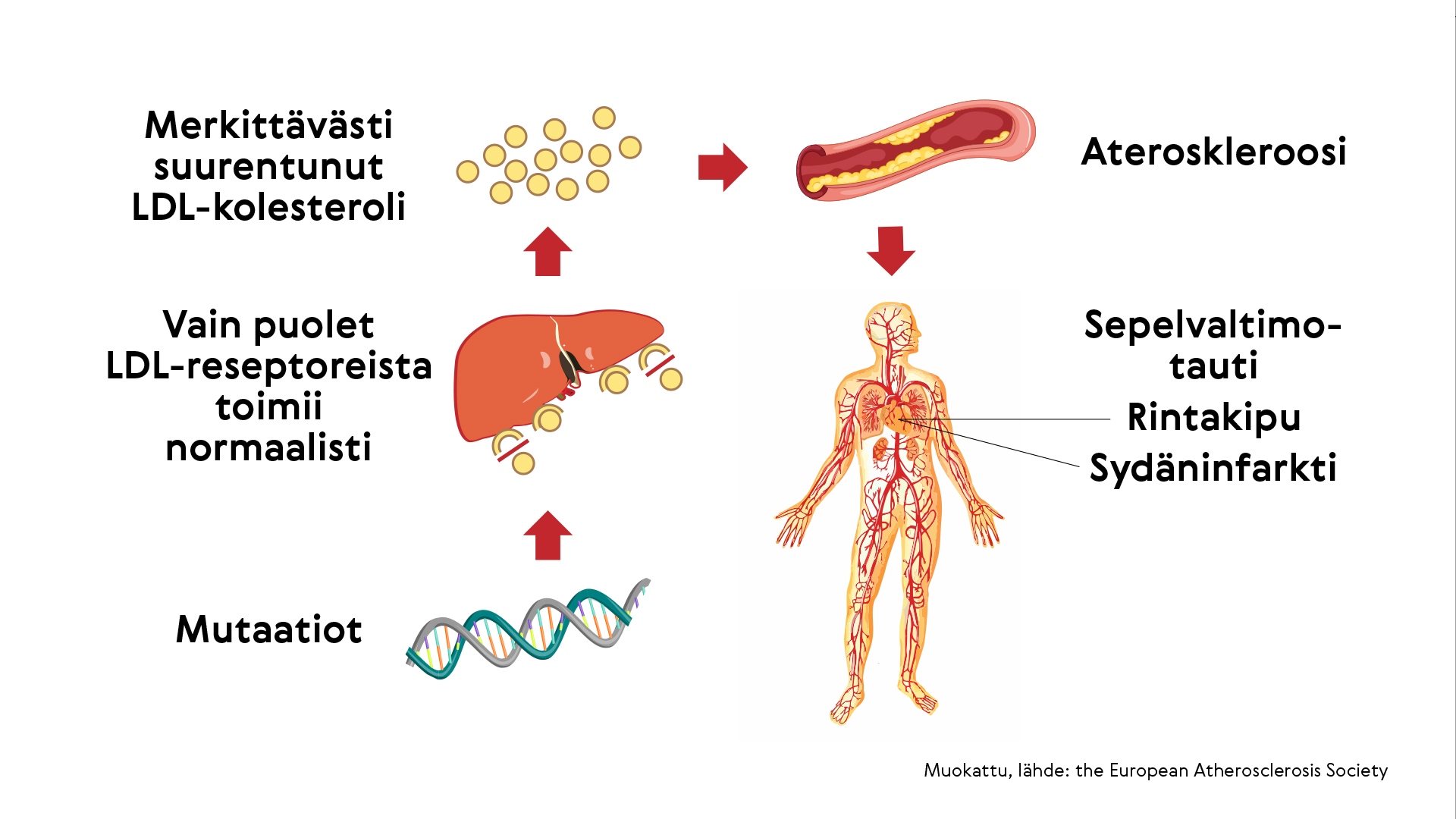
FH:ssa taudin syntytapa on yksinkertainen. Kolesteroli poistuu verestä pääosin siten, että sitä kuljettavat LDL-hiukkaset otetaan maksaan, joka prosessoi ne edelleen. Maksan pinnalla on tarttumakohtia, LDL-reseptoreja, joihin LDL-hiukkaset sopivat pinnallaan olevan apoB-proteiinin välityksellä kuin avain lukkoon. ”Avain” ja ”lukko” kaapataan yhdessä maksasolun sisään.
FH on useimmiten LDL-reseptorin vika. Reseptorit joko puuttuvat tai ne toimivat vajavaisesti. Harvinaisempia FH:n syitä ovat 1) poikkeava apoB, joka ei pysty tarttuman reseptoriin, ja 2) vika LDL-reseptorin kierrätyksessä takaisin maksasolun pintaan (PCSK9-proteiinin liikatoiminta).
FH on periytyvä eli geneettinen sairaus
Toimivat LDL-reseptorit puuttuvat siksi, että niitä koodaava geeni on rakenteeltaan virheellinen eli siinä on haitallinen mutaatio. Kyse on siis geneettisestä eli periytyvästä eli familiaalisesta (suvuttaisesta) sairaudesta.
FH on periytymistavaltaan yksinkertainen. Kuten kaikkia geenejä, myös LDL-reseptorigeenejä on soluissamme kaksi kopiota, toinen isältä ja toinen äidiltä peritty. Normaalitila on se, että kumpikin vastingeeni tuottaa toimivaa LDL-reseptoria.
Kun jommaltakummalta vanhemmalta on peritty viallinen geeni ja toiselta normaali, kyseessä on heterotsygoottinen FH. LDL-reseptorien määrä on vain puolet normaalista. Sen esiintyvyys väestössä on perinteisen käsityksen mukaan 1:500. Yli viiden miljoonan suomalaisen keskuudessa on siis runsaat 10 000 heterotsygoottia. Uusimpien tutkimusten mukaan määrä on suurempi, jopa 1:200.
Jos on perinyt molemmilta vanhemmilta viallisen geenin, kyseessä on homotsygoottinen FH. Kunnollisesti toimivia LDL-reseptoreja ei ole lainkaan. Näin huono onni on vain yhdellä miljoonasta eli Suomessa tällaisia on vain kourallinen.
FH on kolesterolitauti
FH:n tärkein merkkiominaisuus on kokonais- ja LDL-kolesterolin suuri pitoisuus veressä. Heterotsygoottien kokonaiskolesteroliarvot ovat tyypillisesti 8–15 mmol/l, homotsygoottien 12–30 mmol/l.
Kolesteroli tunkeutuu valtimoiden seinämiin käynnistäen suonten ahtautumisen. Osalla FH-potilaista syntyy näkyviä kolesterolikertymiä jänteisiin (jänneksantoomat), tyypillisimmin akillesjänteisiin. Toinen kertymispaikka on silmän sarveiskalvon ulkoreuna, jossa vaalea kehä (arcus corneae) varhaisessa keski-iässä voi olla vihje FH-taudista.
FH altistaa valtimosairaudelle
FH:n merkitys perustuu siihen, että veren suuri kolesterolipitoisuus lisää sepelvaltimotaudin riskin yli 10-kertaiseksi normaaliväestöön verrattuna. 50–50-säännön mukaan heterotsygoottimiehistä 50 % sairastuu sepelvaltimotautiin 50 ikävuoteen mennessä, naiset jonkin verran myöhemmin.
FH on voimakas valtimotaudin riskitekijä kahdesta syystä. Ensinnäkin kolesteroliarvo on yleensä poikkeuksellisen suuri. Toiseksi perinnöllisenä tautina FH ilmenee eli kolesteroliarvot nousevat jo varhaislapsuudessa. Muut kohonneen kolesterolin muodot ilmenevät yleensä vasta nuoressa aikuisiässä, joten FH:ssa on noin 20 vuoden ”etumatka” taudin kehittymisessä. FH-lapsilla voidaankin säännönmukaisesti todeta alkavia valtimomuutoksia, vaikka oireita antava tauti kehittyykin heterotsygooteilla vasta aikuisiässä.
Riskiä suurentaa entisestään, jos FH-potilaalla on muita valtimosairauksien riskitekijöitä: tupakointi, kohonnut verenpaine, liikapaino tai diabetes. Valtimotaudeille on myös geneettisiä suojatekijöitä, joiden turvin jotkut selviävät suhteellisen pitkään sairastumatta.
Harvinaisessa homotsygoottisessa muodossa sepelvaltimotauti on tavallinen jo lapsilla ja nuorilla.
Miten FH todetaan?
Nopea tapa selvittää FH-taudin todennäköisyyttä on tehdä FH-testi. Testin taustalla olevia muuttujia kuvataan seuraavassa.
Uuden FH-diagnoosin lähtökohtana on edellä mainituissa lukemissa oleva kolesteroliarvo. Pelkkä kokonaiskolesteroliarvon määritys ei riitä, vaan on tutkittava kaikki ns. veren lipidit: kolesteroli, HDL-kolesteroli, LDL-kolesteroli ja triglyseridit. Sudenkuoppana voi olla huomattavasti kohonnut triglyseridiarvo, jonka seurauksena myös kolesteroliarvo voi nousta FH:lle ominaisiin lukemiin.
Jos geenitestiä ei tehdä tai sen tulos on epävarma, voidaan käyttää taulukon mukaista kliinistä pisteytystä.
| Ominaisuus (vain yksi, suurimmat pisteet antava, ominaisuus kustakin ryhmästä otetaan huomioon) | Pisteet |
|---|---|
| Ryhmä 1: Sukutausta | |
| Ensimmäisen asteen sukulaisella varhainen (miehellä < 55-v, naisella < 60-v) sepelvaltimotauti | 1 |
| tai | |
| Vähintään 18-vuotiaan ensimmäisen asteen sukulaisen LDL-kolesteroli > 5 mmol/l | 1 |
| Ensimmäisen asteen sukulaisella jänneksantoomia ja/tai sarveiskalvon ”arcus” | 2 |
| tai | |
| Alle 18-vuotiaan lapsen LDL-kolesteroli > 4 mmol/l | 2 |
| Ryhmä 2: Tutkittavan taustatiedot | |
| Tutkittavalla varhainen (miehellä < 55-v, naisella < 60-v) sepelvaltimotauti | 2 |
| tai | |
| Tutkittavalla varhainen (miehellä < 55-v, naisella < 60-v) aivo- tai ääreisvaltimotauti | 1 |
| Ryhmä 3: Tutkittavan löydökset | |
| Jänneksantoomia | 6 |
| Sarveiskalvon ”arcus” < 45-v:na | 4 |
| Ryhmä 4: Tutkittavan LDL-kolesteroliarvo (mmol/l) | |
| Yli 8,5 | 8 |
| 6,5–8,4 | 5 |
| 5,0–6,4 | 3 |
| 4,0–4,9 | 1 |
| Ryhmä 5: Geenitesti | |
| FH-tautia aiheuttava mutaatio | 8 |
*Jakauma on ikä-, sukupuoli- ja maakohtainen
Tulkinta:
- Varma FH: > 8 pistettä
- Todennäköinen FH: 6–8 pistettä
- Mahdollinen FH: 3–5 pistettä
- Todennäköisesti ei FH: 0–2 pistettä
Selväpiirteisin tapa todeta FH on geenitesti. Kuten taulukostakin ilmenee, FH:lle ominaisen mutaation löytyminen antaa käytännössä varman diagnoosin.
Geenitesti
Geenitesti on syytä tehdä, mikäli mahdollista, FH-diagnoosin varmistamiseksi ja sukuselvityksen pohjaksi.
FH:ta aiheuttavia geenimutaatioita on tuhansia. Suomessa tilanne on yksinkertaisempi, koska väestömme geneettisen rakenteen vuoksi noin 80 % FH-tapauksista johtuu jostain kahdeksasta valtamutaatiosta (FH-Helsinki, FH-Pohjois-Karjala, FH-Turku, FH-Pori, FH-Pogosta, FH-Keuruu, FH-Espoo ja FH-Pro84Ser).
Jos suvussa oleva mutaatio tunnetaan, tämän geenivirheen testaaminen sukulaisilta on melko edullista (nykyään noin 200 euroa).
Jos etsittävää geenivirhettä ei tiedetä, voidaan joutua laajempiin ja kalliimpiin (1 000–1 200 euroa) geenitutkimuksiin.
Joskus geenivirhettä ei laajassakaan tutkimuksessa löydy, vaikka kliininen pisteytys viittaisi FH-tautiin. Tällöin kyseessä on todennäköisesti usean kolesteroliarvoa heikommin nostavan geenin yhteisvaikutus, eräänlainen negatiivinen lottovoitto. Kyseisen yksilön hoidon kannalta tällä ei ole merkitystä (hän on kolesterolia alentavan hoidon tarpeessa), mutta sukuselvitystä geenitestillä ei kannata tehdä.
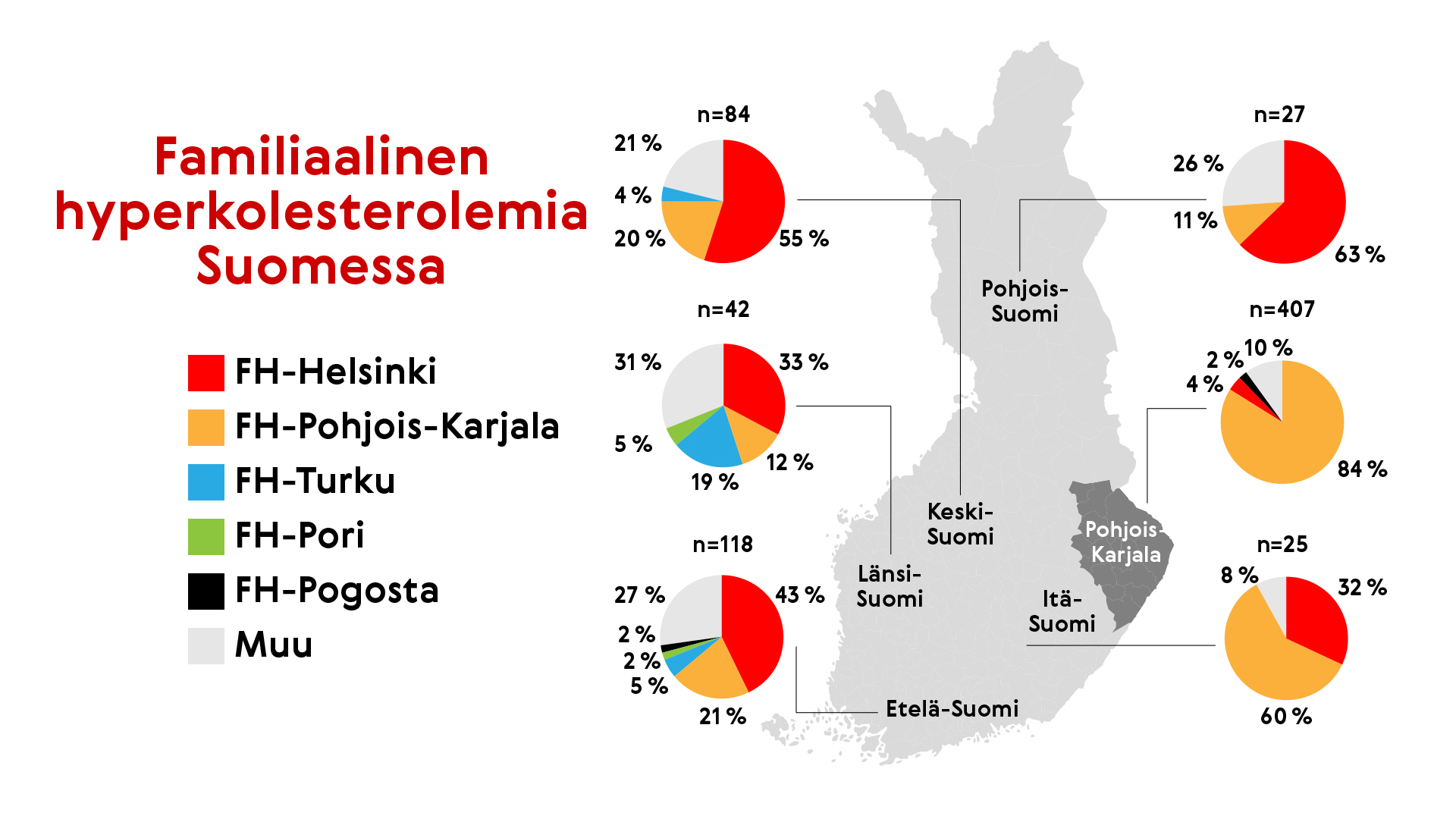
Suomessa esiintyviä FH-mutaatioita on useita.
Suvun tutkiminen
Tehokkain keino saada FH-potilaat nykyistä paremmin hoidon piiriin on sukupolviseulonta (engl. cascade screening).
Tavallisin lähtökohta on FH-potilas, jonka mutaatio on tiedossa. Tällöin hänen elossa olevilta ensimmäisen asteen sukulaisiltaan (vanhemmat, sisarukset, lapset) tutkitaan kyseinen mutaatio (esim. FH-Helsinki tai FH-Pohjois-Karjala). Tästä edetään löytyneiden uusien FH-potilaiden sukulaisiin jne. Vaihtoehtoisesti voidaan tutkia myös kolesteroliarvot (erityisesti LDL-kolesteroli), ellei geenitestiä ole helposti saatavissa.
Toinen mahdollisuus on kliininen FH-potilas, jonka geenejä ei ole tutkittu tai laajassakaan geenitestissä ei ole löydetty mutaatiota. Tällöin vastaava sukututkimus tehdään käyttäen LDL-kolesterolimittausta.
Toistaiseksi harvinainen tilanne on se, että henkilöllä on tunnistettu FH-tautiin liitetty geenivirhe, mutta hänellä ei ole kliinistä FH-tautia. Tällöin hänen ja hänen sukulaistensa LDL-kolesterolia tulee seurata.
Lapsille sukupolviseulonta suositellaan tehtäväksi 2–10 vuoden iässä.
FH:n hoito
Kaikki FH-potilaat tarvitsevat elintapahoitoa, joka tähtää siihen, ettei geneettisen haitan lisäksi ole muita valtimosairautta jouduttavia riskitekijöitä. Ruokavalioksi suositellaan tavanomaista sydänystävällistä ravintoa. Kolesteroliarvojen kannalta tärkeintä on rasvan laatu: pehmeän (tyydyttymättömän) rasvan suosiminen kovan (tyydyttyneen) kustannuksella. Tärkeää on myös välttää liikapainoa ja verenpainetta nostavaa liikaa suolaa. Tupakoimattomuus on ehdottoman tärkeää. Liikunnallinen elämäntapa on monin tavoin terveyttä tukevaa. Verenpainetta on syytä seurata ja tarvittaessa hoitaa.
FH-potilas tarvitsee elintapojen lisäksi myös kolesterolilääkitystä. Peruslääke on statiini. FH:ssa on tarpeen käyttää tehokasta statiinia maksimiannosta (atorvastatiini 80 mg/vrk tai rosuvastatiini 40 mg/vrk) tai, jos näistä aiheutuu haittavaikutuksia, suurinta siedettyä annosta. Hoitotavoitteisiin pääsemiseksi tarvitaan usein yhdistelmähoitoa, jolloin statiinin rinnalle liitetään kolesterolin imeytymistä suolistosta estävä etsetimibi.
Lisääntymisikäisten naisten kohdalla on muistettava, että statiineja ei voi käyttää raskauden aikana.
Lapsilla on tutkimustuloksia, joiden mukaan statiineja voidaan turvallisesti käyttää 8-vuotiaasta lähtien. Tämän on tapahduttava hoitoon erityisesti perehtyneen lastenlääkärin valvonnassa. FH-diagnoosi on hyödyllistä saada jo ennen lääkehoitoikää, jotta ruokavalio ja muut terveelliset elintavat tulevat luontevaksi osaksi elämää alusta alkaen.
Uusin lääkeaineryhmä hyperkolesterolemian hoidossa ovat PCSK9-proteiinin vasta-aineet. Ne vaikuttavat lisäämällä LDL-reseptorien kiertoa takaisin maksasolun pintaan sen jälkeen, kun ne ovat purkaneet LDL-lastinsa soluun. Maksan LDL-reseptorien määrä siis lisääntyy ja kolesterolin poistuminen verenkierrosta maksaan tehostuu. Suomessa on saatavana kaksi tämän ryhmän lääkeainetta, evelokumabi ja alirokumabi. Lääkkeet pistetään ihon alle 2–4 viikon välein. Niitä voi käyttää statiinin sijasta, jos statiini ei sovi, tai statiinin rinnalla hoidon tehostamiseksi. Sama vaikutus saadaan kolmannella, puolen vuoden välein pistettävällä inklisiraani-valmisteella. Lääkkeet ovat lääkärin B-lausunnolla rajoitetusti peruskorvattavia FH-potilaille.
Hoidolla lasketaan LDL-kolesterolia. Tavoite on aikuisilla alle 1,8 mmol/l ja lapsilla alle 3,5 mmol/l. Jos henkilö on jo sairastunut valtimotautiin, esim. sepelvaltimotautiin, tavoite on alle 1,4 mmol/l.
Harvinaiseen homotsygoottiseen FH-tautiin ainoa tehokas hoito toistaiseksi on ollut työläs ja kallis LDL-afereesi, jossa verta puhdistetaan kierrättämällä se kehon ulkopuolella laitteen läpi, joka poistaa siitä LDL-hiukkasia. Homotsygoottiseen FH-tautiinkin on tulossa uusia lääkkeitä.